Introduzione
L’amiloidosi è una malattia sistemica, cioè una patologia in grado di coinvolgere potenzialmente più organi, contemporaneamente o in tempi diversi. È inoltre una malattia infiltrativa, ovvero caratterizzata dalla deposizione di organi e tessuti da parte di materiale insolubile, le fibrille, composte principalmente da proteine. Questi precursori proteici, originalmente solubili, nel corso della vita vanno incontro a cambiamenti conformazionali, ereditari o acquisiti, in grado di favorirne l’aggregazione e quindi la precipitazione.
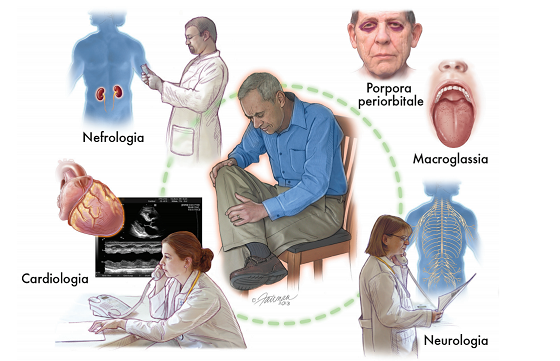
Classificazione
Sono note più di trenta forme d’amiloidosi, classificate in base alla proteina precursore. Tuttavia, la grande maggioranza delle amiloidosi a coinvolgimento cardiaco sono riconducibili a sole tre forme:
– l’amiloidosi AL, nelle quali una porzione degli anticorpi (le catene leggere delle immunoglobuline) viene prodotta in forma ripetitiva ed alterata da parte di cellule deputate a ciò (plasmacellule), come avviene nel corso di malattie onco-ematologiche quale il mieloma multiplo;
– l’amiloidosi ATTR, nelle quale una proteina prodotta in gran parte dal fegato, la transtiretina, può essere alterata a causa di mutazioni geniche (forma ATTR ereditaria) o alterazioni acquisite con l’età (forma ATTR wild-type).
Le tre forme sono tutte caratterizzate dalla progressiva infiltrazione di tutte le strutture cardiache (muscolo cardiaco, valvole, pericardio). L’infiltrazione del muscolo cardiaco determina una maggiore rigidità del cuore il quale, normalmente, si comporta come una pompa elastica, in grado cioè di distendersi per accogliere il sangue di ritorno dalle vene e successivamente di contrarsi per eiettare il sangue attraverso il circolo arterioso. La rigidità acquisita nel corso dell’amiloidosi comporta da un lato l’incapacità del cuore a maneggiare il ritorno venoso e dall’altro la difficoltà nel portare il sangue arterioso all’organismo per assicurare la corretta funzione degli organi. Ne consegue un accumulo di liquidi a monte rispetto al cuore e una scarsa irrorazione periferica a valle dello stesso che giustificano l’insorgenza di sintomi e segni (scompenso cardiaco) quali la fame d’aria, gli edemi periferici e l’astenia. Inoltre il coinvolgimento delle vie di conduzione dell’impulso cardiaco è in grado di determinare l’insorgenza di aritmie quale la fibrillazione atriale e il blocco della conduzione stessa a vari livelli del cuore (blocco seno-atriale, blocco atrio-ventricolare, blocchi di branca). Ancora, il coinvolgimento e il rimaneggiamento degli apparati valvolari può determinarne l’incapacità di contenere il flusso ematico in senso retrogrado (insufficienza) o di assicurare un adeguato flusso anterogrado (stenosi). Classica l’associazione, specie nelle forme ATTR wild-type, dell’amiloidosi cardiaca con la stenosi della valvola aortica.
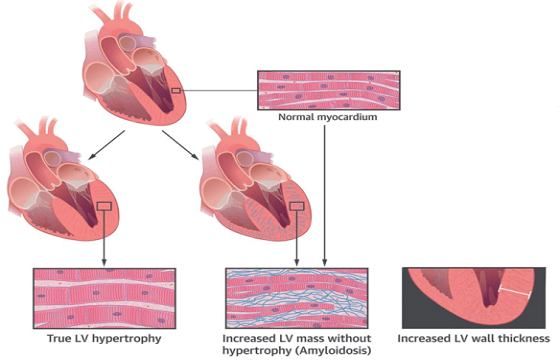
Clinica
Le manifestazioni cardiache d’amiloidosi sono piuttosto aspecifiche e ciò giustifica in parte la possibilità di confondere una malattia piuttosto rara quale l’amiloidosi con malattie cardiache più comuni, specie se la valutazione dei pazienti avviene in centri non specializzati, favorendo in tal modo la mancata diagnosi o il ritardo della stessa specie nelle prime fasi della malattia, laddove ci sarebbe maggior margine per l’efficacia dei trattamenti ad oggi disponibili. Nonostante il coinvolgimento cardiaco e le conseguenti manifestazioni cliniche sia sostanzialmente sovrapponibile tra le forme su citate, tuttavia la storia naturale varia notevolmente tra di esse. Le forme AL sono quelle caratterizzate da maggiore mortalità, mentre le forme ATTR, specie a seguito dell’introduzione di nuove terapie specifiche, hanno un decorso sostanzialmente più benigno. Ciò si traduce nella necessità di diagnosi quanto più precoci e definite possibili.
Diagnosi
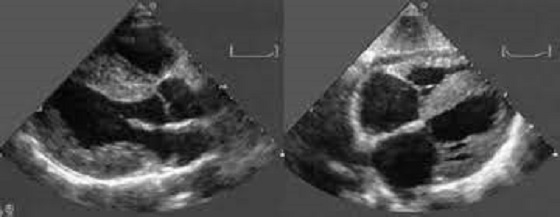
Negli ultimi anni gli avanzamenti nella conoscenza della malattia hanno portato ad un incremento costante di diagnosi di amiloidosi cardiaca. L’imaging cardiovascolare, consistente nell’esecuzione di un esame ecocardiografico transtoracico o di una risonanza magnetica nucleare cardiaca, consente spesso di far sorgere un nuovo sospetto diagnostico o di confermare un sospetto sorto sulla scorta della storia clinica del paziente o di altre metodiche diagnostiche. Tali metodiche, che hanno il vantaggio di essere non invasive e di facile esecuzione (specie l’ecocardiografia), hanno consentito di affinare l’abilità diagnostica in ambito cardiologico. Tuttavia esse da sole non consentono una diagnosi di certezza della malattia ma consentono solo di intraprendere il percorso diagnostico corretto per giungere alla diagnosi di certezza dell’amiloidosi. Anche in questo campo si sono registrati numerosi avanzamenti. Basti pensare che fino a pochi anni fa la diagnosi d’amiloidosi era solo bioptica, cioè richiedeva il prelievo degli organi e dei tessuti colpiti in modo invasivo, cosa che rendeva in processo diagnostico più macchinoso. Ad oggi, almeno per la forma ATTR, è possibile una diagnosi non invasiva nel caso in cui si sia esclusa una forma AL con un semplice prelievo ematico e un esame specifico sulle urine (immunofissazione sierica ed urinarie delle immunoglobuline e determinazione delle catene libere leggere nel siero) e si confermi il coinvolgimento cardiaco con un esame semplice e a basso costo quale la scintigrafia ossea, grazie alla capacità dell’amiloide di captare con elevata avidità il composto osseo-specifico.
Trattamento
La forma AL è sostanzialmente una malattia ematologica con manifestazioni anche cardiache e il trattamento si basa su chemioterapie specifiche volte alla soppressione della produzione di anticorpi anomali da parte del midollo osseo. Le forme ATTR hanno invece manifestazioni perlopiù neurologiche e cardiologiche e i nuovi trattamenti, da poco disponibili, si basano sull’interruzione dei fenomeni di fibrillogenesi, ovvero quei processi in grado di tramutare precursori proteici solubili in composti insolubili componenti le fibrille. Sempre per la forma ATTR con coinvolgimento cardiaco è ad oggi disponibile una molecole, il tafamidis, un farmaco che agisce da stabilizzatore del precursore proteico, la transtiretina nella sua forma originale di tetramero indissociato, che si è dimostrato in grado di migliorare la qualità di vita e di aumentare la sopravvivenza nei soggetti trattati. Sono inoltre in corso trials clinici che stanno valutando l’impatto sul benessere e la sopravvivenza in pazienti con manifestazioni cardiologiche con molecole ad azione genica, rivoluzionarie in questo ambito, quali il patisiran e l’inotersen che hanno già dimostrato la capacità di migliorare la clinica dei soggetti con amiloidosi con manifestazioni neurologiche.
Conclusioni
In conclusione, negli ultimi anni tutti i preconcetti sull’amiloidosi cardiaca, considerata erroneamente estremamente rara, difficile da diagnosticare e sostanzialmente intrattabile, sono stati sconfessati. Alle maggiori conoscenze sulle dinamiche della malattia si sono associate nuove strategie diagnostiche e terapeutiche che gettano una luce di speranza sul futuro dei pazienti affetti dalla malattia.
Contatta l’esperto riguardo a quest’argomento
Dott. Giuseppe Palmiero
Dirigente Medico, U.O.C. Cardiologia
Unità di Malattie Genetiche e Rare Cardiovasculari, Centro di Coordinamento Malattie Rare
AORN dei Colli – Ospedale Monaldi – Napoli
Dott.ssa Marta Rubino
Unità di Malattie Genetiche e Rare Cardiovasculari, Centro di Coordinamento Malattie Rare
AORN dei Colli – Ospedale Monaldi – Napoli
Dott.ssa Erica Vetrano
Unità di Malattie Genetiche e Rare Cardiovasculari, Centro di Coordinamento Malattie Rare
AORN dei Colli – Ospedale Monaldi – Napoli